Balto: AI-Powered Computational Drug Discovery & Molecular Modeling Platform
1. Overview
Balto is an AI-powered computational assistant developed by Deep Origin to accelerate drug discovery and democratize access to in silico molecular modeling tools. It provides a robust suite of functionalities, including:
- Molecular Docking: Simulate the interaction of ligands (small molecules) with protein targets
- Molecular Property Prediction: Predict key properties like lipophilicity, solubility, and other ADMET properties
- Protein-Ligand Interaction Analysis: Analyze the results of docking simulations, identifying key protein-ligand interactions across different binding poses.
- Data Search: Find information about ligands and proteins and their homologs- from multiple public databases (PDB, ChEMBL, UniProt, PubChem, BindingDB, AlphaFold, Open Targets)
- Data Processing: Handle and analyze molecular datasets, including CSV files
- Visualization: Generate interactive 2D and 3D visualizations of molecules, proteins, and docking results and edit in our 2D chemical structure editor.
2. Key Features
Balto offers a wide range of features to support drug discovery.
2.1 Molecular Data Retrieval & Informatics
Balto integrates with several key databases, allowing you to retrieve comprehensive molecular and protein data:
- Protein Data Bank (PDB): https://www.rcsb.org/ – Retrieve protein structural data
- ChEMBL: https://www.ebi.ac.uk/chembl/ – Access information on bioactive molecules, drug targets, and assay data
- UniProt: https://www.uniprot.org/ – Search protein sequences and annotations
- PubChem: https://pubchem.ncbi.nlm.nih.gov/ – Query information on small molecules and bioassay data
- BindingDB: https://bindingdb.org/rwd/bind/index.jsp – Retrieve protein-ligand affinity data
- Open Targets: https://www.opentargets.org/ - Query pre-competitive insights into drug target selection
- Homology search - Find proteins based on sequence similarity
- Patent Check – Identify patented molecules by inputting a SMILES string or a list of SMILES strings to check for existing patents
- Analyze PDF Documents – Extract valuable insights from scientific publications, including summarizing key findings, identifying important data points, and retrieving relevant sections
- Convert Between Molecular Identifiers – Facilitate conversion between different molecular representations such as SMILES strings, CAS numbers, and IUPAC names to ensure consistency across chemical databases
2.2 Molecular Property & ADMET Profiling
Balto provides in silico predictions for key molecular properties and toxicity assessments, helping you evaluate the drug-likeness of compounds:
- Physicochemical Properties:
- LogP: Estimate lipophilicity
- LogD: Calculate the pH-dependent distribution coefficient
- LogS: Predict aqueous solubility
- Toxicity Predictions:
- hERG Inhibition Prediction: Assess the risk of cardiotoxicity
- CYP450 Interaction Prediction: Analyze metabolic stability
- Ames Mutagenicity Estimation: Model the risk of carcinogenicity
- Aggregated Theoretical Descriptors:
- Synthetic Accessibility Score (SAS): Assess molecular complexity
- Quantitative Estimate of Drug-likeness (QED): Evaluate overall drug-likeness
2.3 Protein Structure Analysis & Ligand Preparation
Balto offers tools for manipulating protein structures and preparing ligands for docking:
- Protein Processing & Structural Manipulations:
- Binding Pocket Identification: Find potential binding sites on a protein
- Protein Structure Alignment: Align multiple protein structures and find mutation spots
- Find homology analog: Find protein homolog and highlight sequence difference if protein structure of your target is not available
- Ligand Preparation:
- Protonation State Prediction: Determine the likely protonation state of a ligand at a given pH
- Functional Group Identification: Identify functional groups within a molecule
- Molecular Similarity Search: Find molecules similar to a query molecule
2.4 Molecular Docking
Balto supports rigid ligand docking and subsequent analysis:
- Rigid Docking: Perform molecular docking simulations into a rigid pocket
- Docking Analysis: Analyze the results of docking simulations, including binding energies and key protein-ligand interactions
- Docking Box Visualization: Visualize the defined docking box in 3D
- Ligand and Protein Preparation: Tools for preparing ligands and proteins for the docking process
2.5 Data Processing & Analysis
Balto automates data handling and analysis:
- Molecular Dataset Handling:
- CSV Data Extraction, Processing, and Querying:
- Data Extraction: Read and extract molecular data from CSV files containing SMILES strings, molecular properties, or assay results
- Data Processing: Perform cleaning, handle missing values, and transform the dataset into a suitable format for analysis
- Querying: Filter and query CSV data using SQL-like commands to extract specific rows, columns, and aggregated insights
- Summarize your lead series data: Provide activity data with SMILES, calculate ADME properties and let Balto recommend your top molecules with balanced properties
- Batch Data Processing & Molecular Analysis Pipelines:
- Batch Processing: Handle multiple molecules simultaneously instead of one-by-one, improving efficiency for large datasets
- Molecular Analysis Pipelines: Automate workflows for molecular screening, such as batch docking, property prediction, and dataset filtering
- Batch Docking: Perform docking on multiple molecules at once, reducing computational time and increasing throughput for drug discovery applications
- CSV Data Extraction, Processing, and Querying:
- Computational Workflow Automation:
- File Management and Processing: List available files in the session’s directory to identify accessible data; Retrieve and stream file content for analysis without separate downloads.
- Basic Retrosynthetic Engine: Predict basic synthetic routes for complex molecules
2.6 Structural & Visualization Tools
Balto provides powerful visualization capabilities:
- Molecular Rendering & Visualization:
- SMILES Structure Drawing: Generate 2D representations of molecules from SMILES strings. Our chemical structure editor allows you to draw and edit molecules visually in a 2D workspace.
- Protein Visualization: Create interactive 3D visualizations of protein structures
- Binding Pocket Visualization: Highlight and visualize identified binding pockets
- Docking Results Visualization: Display docking results interactively, showing ligand poses and interactions
- Structural Analysis:
- Calculate Structural RMSD: Calculate the Root Mean Square Deviation (RMSD) between structures
- Mutagenesis and Homology Analysis: Compare two protein structures to identify mutations and structural gaps
3. Getting Started
- Account Creation/Login: Sign up for an account here or log in to your existing account.
- Select products of interest: Upon logging in, you will land on a Deep Origin’s gateway page where you can select which product you want to interact with
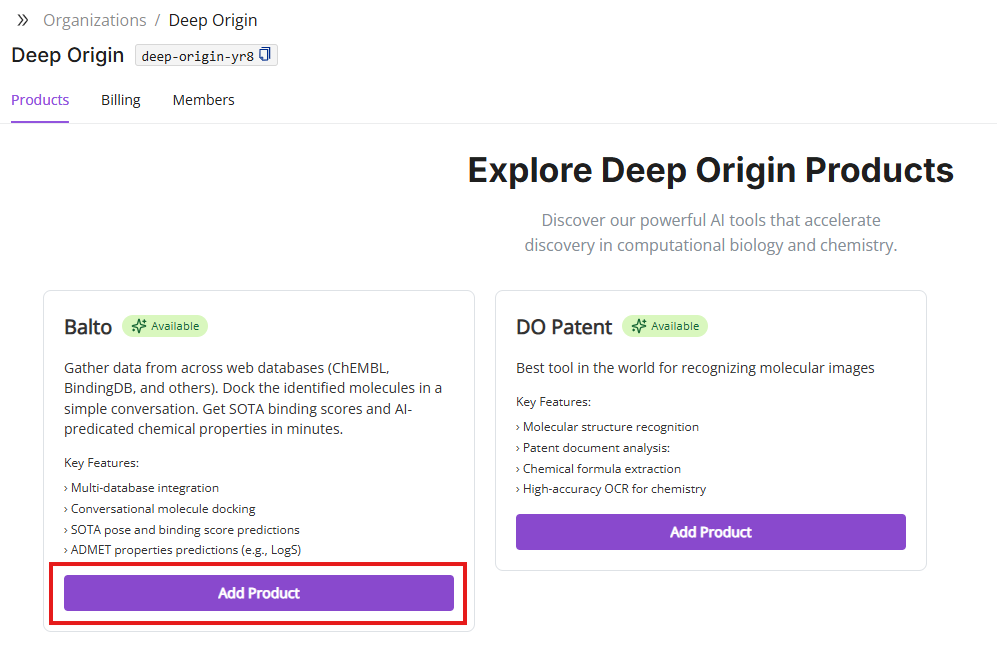
- Initial Balto Interface: Upon logging in, you’ll be presented with the main Balto interface, which is organized into distinct panels (described below).
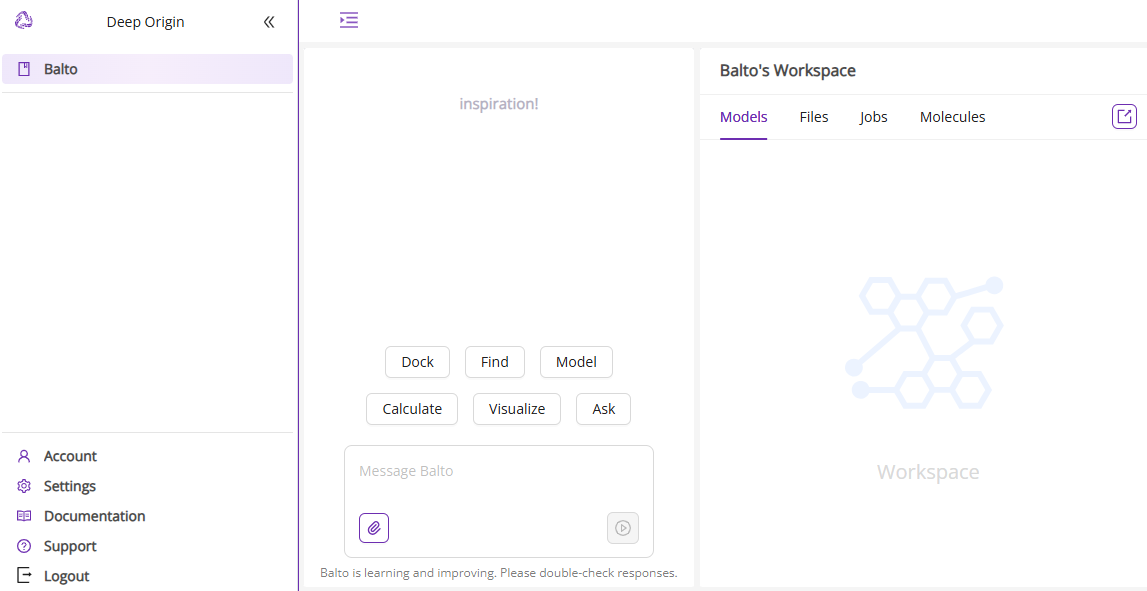
- Access Balto: If the interface above is not what you see, navigate to Balto within the Deep Origin platform from the left panel. You will find it listed among the available products.
4. User Interface (UI)
The Balto interface is designed for intuitive navigation and efficient workflow execution. It is divided into four main panels (see detailed descriptions in the following sessions):
- Products and Account Settings panel
- Chat Space panel
- Balto Workspace panel
- Chat Settings panel
4.1 Products and Account Settings Panel
The left panel provides access to various Deep Origin products and account settings. It is divided into two segments:

Products and Settings
- Top Segment: Displays a list of your active Deep Origin products that you can immediately access via your account (e.g., Balto)
- Bottom Segment: Provides links to:
- Account: Manage your account information (first name, last name, title, company, password). Clicking “Account” takes you to the account settings page.
- Settings: Access Deep Origin’s product selection, pricing & billing details, manage team members. (See Settings Menu for details.)
- Documentation: Direct access to this documentation
- Support: Go to the Deep Origin support portal to request features or report issues
- Logout: Log out of your Balto account
You can collapse or expand this panel by clicking the double arrow (<<) next to your name.
4.2 Chat Space Panel
The middle panel is the primary interaction area where you communicate with Balto using natural language. It displays your current conversation with Balto. When you first log in, this area shows typical tasks that Balto can execute.
On mobile:
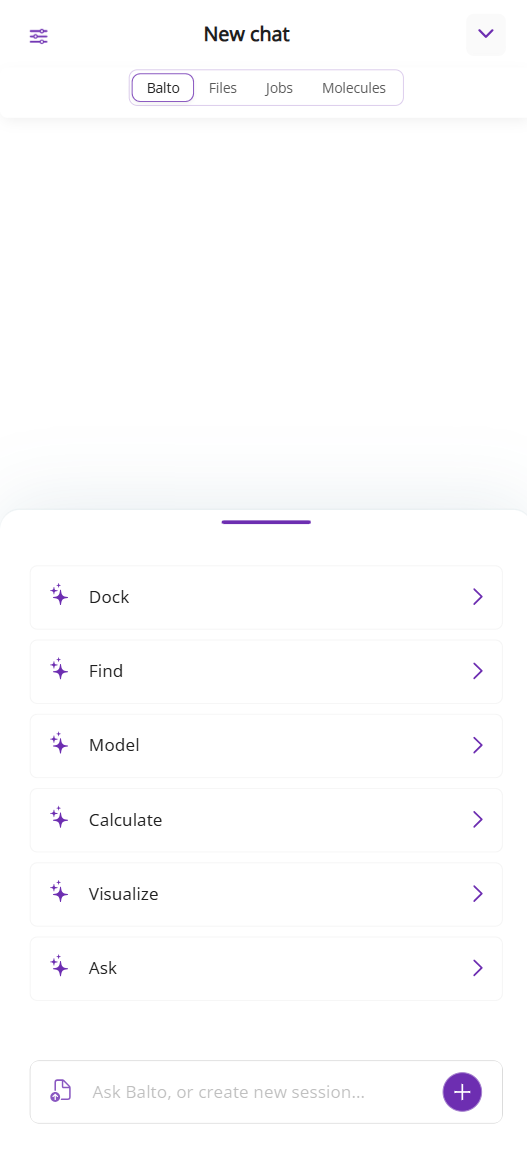
The panel consists of:
- Top Segment: The conversational area displaying your prompts and Balto’s responses.
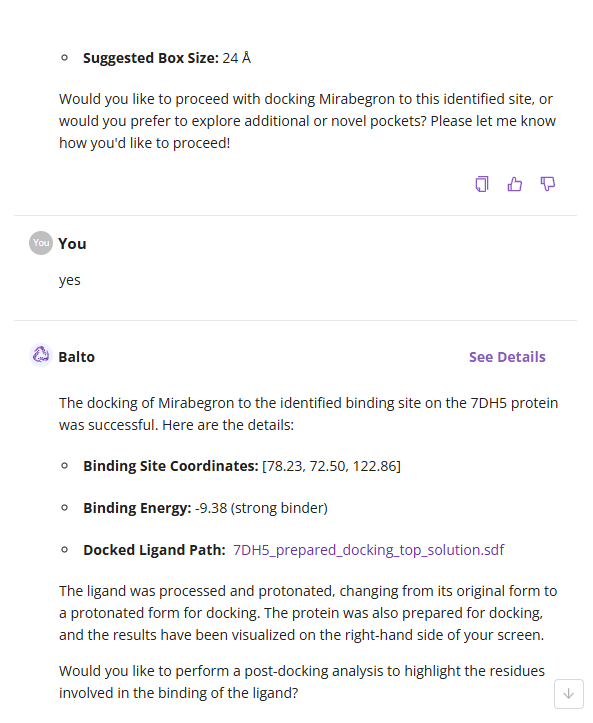
- Middle Segment: Suggested actions that Balto can perform (e.g., Dock, Find, Model, Calculate, Visualize, Ask). Clicking an action provides example prompts.
The default Balto tasks will disappear once you start interacting with Balto.
- Bottom Segment: The input area where you type your requests. You can also:
- Upload files using either the paperclip icon (
📎) or drag-and-dropping files of interest into this area. - Open the 2D chemical structure editor using the Create Molecule icon.
- Select the Research button to prompt a long-form research report about a topic. The report is viewable in the chat as well as in the Results.md file in the Files tab of the Balto Workspace panel.
- Upload files using either the paperclip icon (
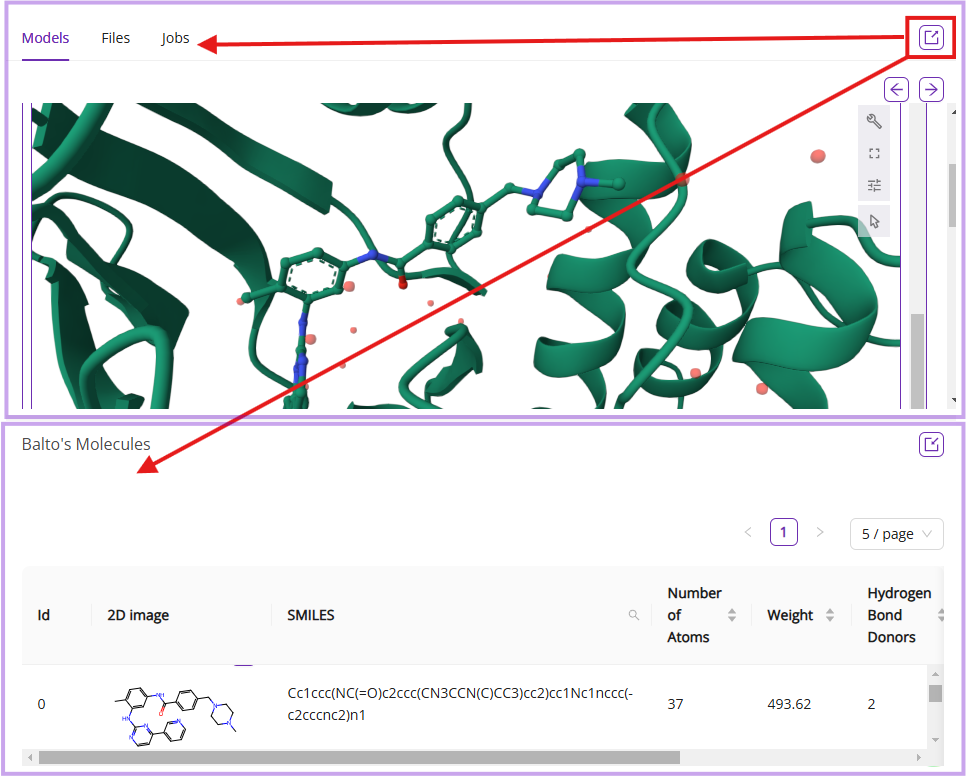
4.3 Balto Workspace Panel
The right panel lets you access data produced by Balto. It includes intermediate files, visualizations, jobs, and more. It has four tabs:
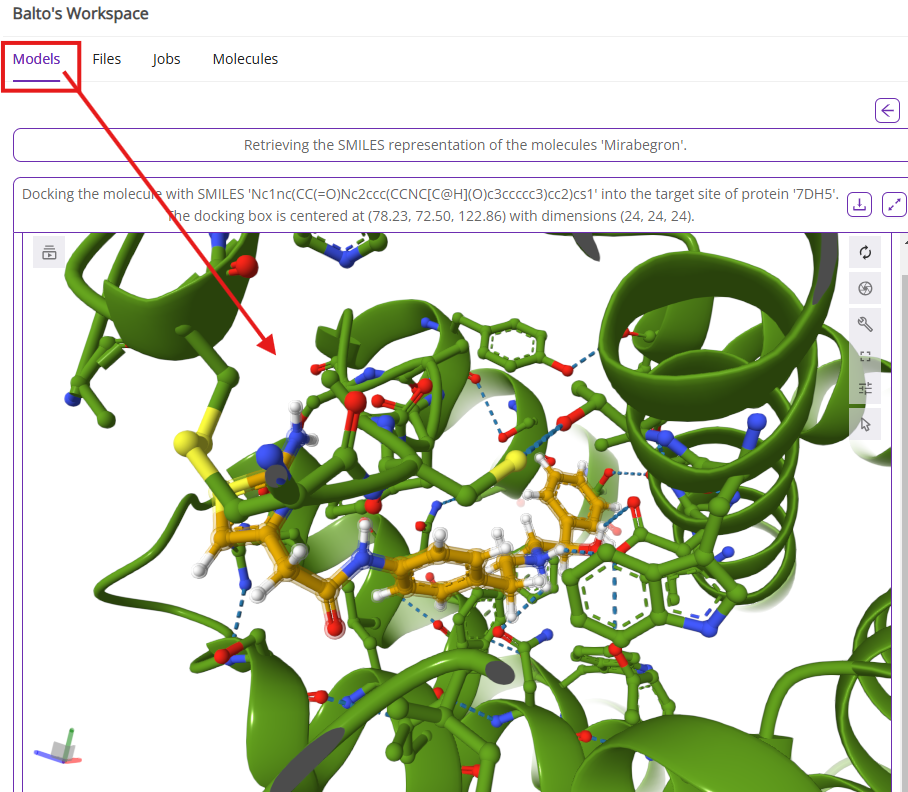
- Models: Displays 3D visualizations generated by Balto (e.g., protein structures, ligand poses) that you can interact with (rotate, zoom, etc.).
- Files: Lists all files uploaded or generated during your session.
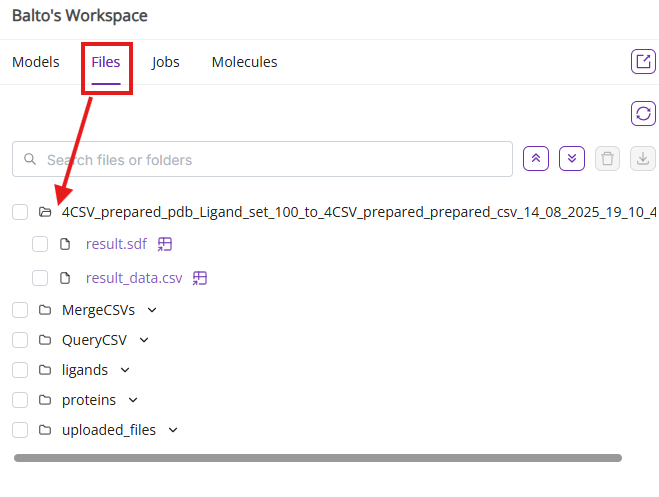
- The folder navigation has necessary navigation to help find and do operations with the files of interest
- Search box

- Expand all and collapse all buttons

- Refresh button

- Delete button (active when file or folder are selected)

- Download button (active when file or folder are selected)

- Search box
- The folder navigation has necessary navigation to help find and do operations with the files of interest
- Jobs: Shows the status of any background tasks (e.g., docking simulations) with progress updates and result links.
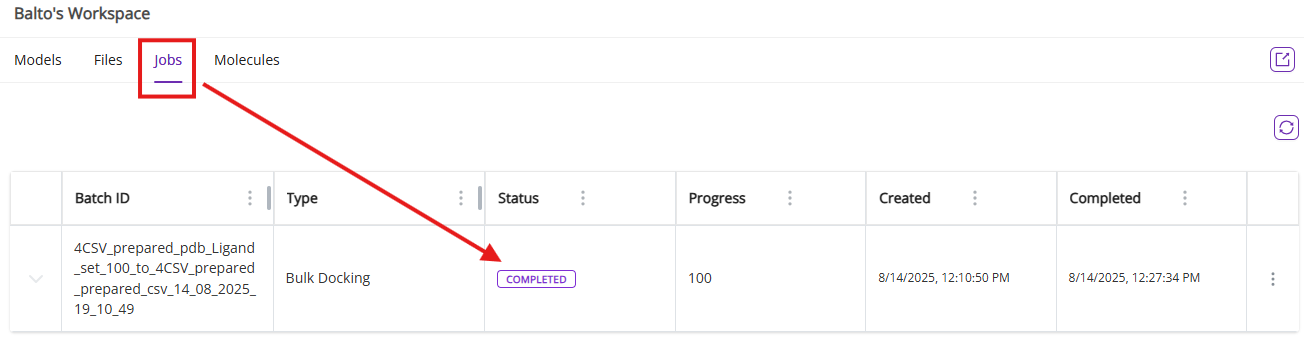
- From jobs menu, you access results files to process them externally by clicking on Open Files
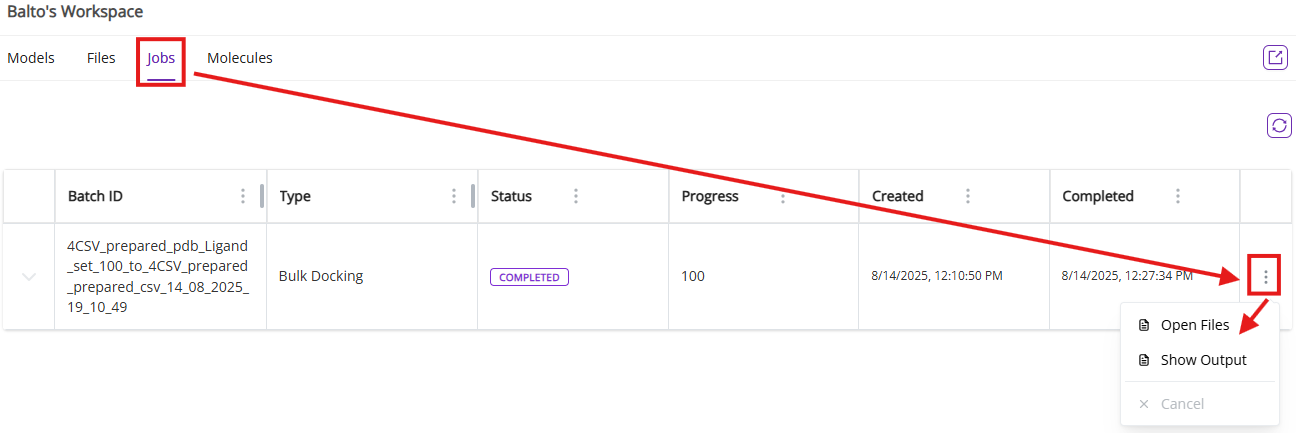
- You can also review docking output in the expanded view by clicking on Show Output
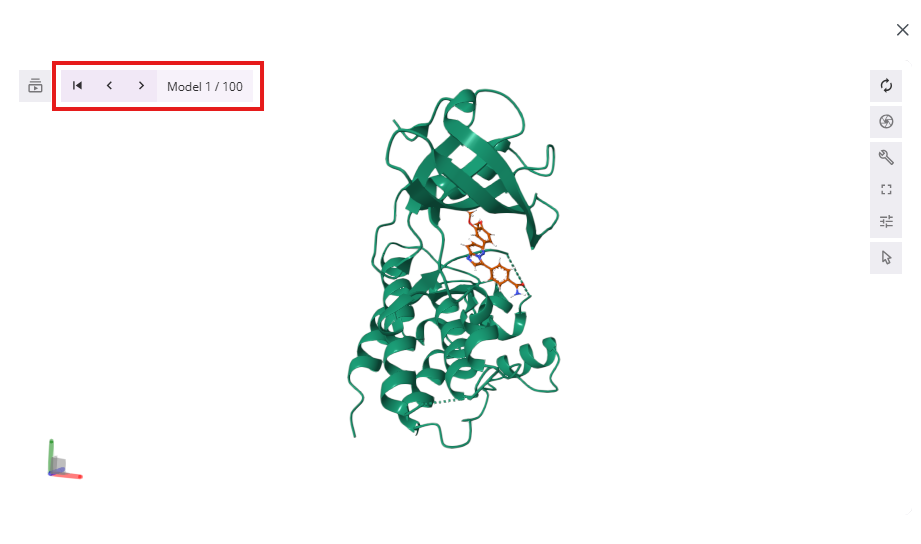
- In the results, window you can manually inspect docked poses by clicking on back and forward buttons
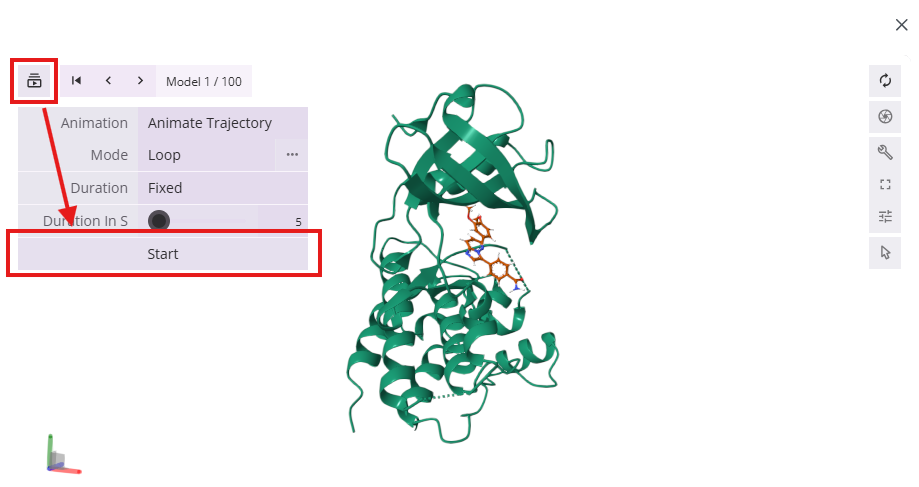
- Additionally, you can quickly review poses in an automatic movie format
- In the results, window you can manually inspect docked poses by clicking on back and forward buttons
- From jobs menu, you access results files to process them externally by clicking on Open Files
- Molecules: Lists all of the molecules gathered or uploaded during the session, displaying 2D structures, SMILES strings, and other properties. You can click on the hyperlinked SMILES string to open the 2D chemical structure editor for that molecule.
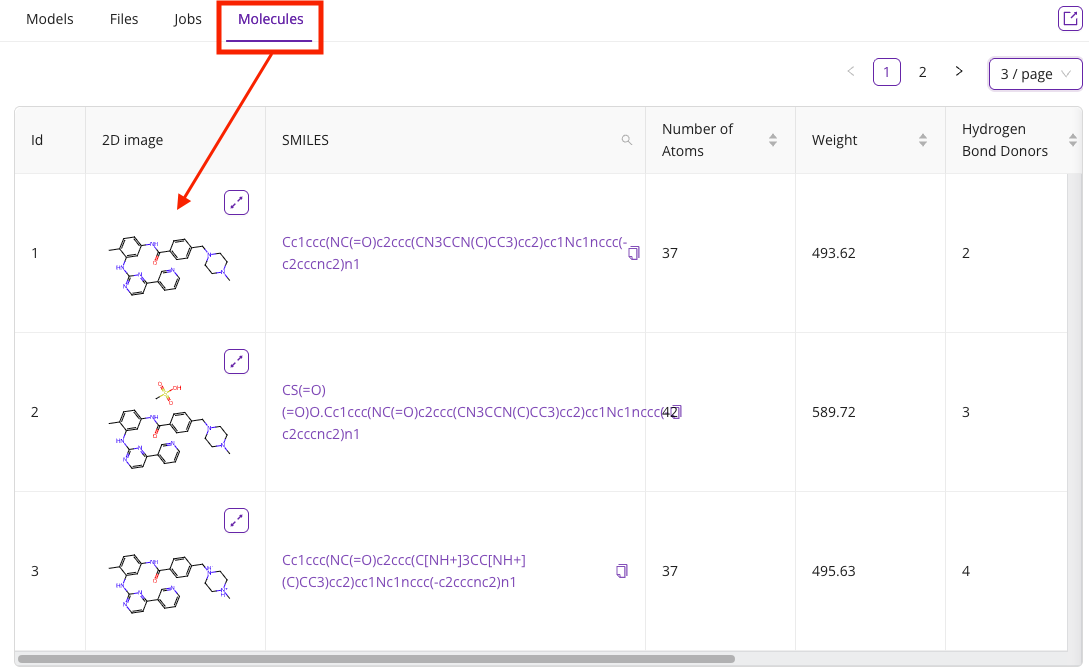
You can extract a tab into a separate segment window by clicking the extract icon:
![]()
On mobile, these tabs are located at the top of the chat
4.4 Chat Configuration Panel
The top panel provides options for managing your current Balto chat:
Menu Button: Shows or hides the Balto chat menu

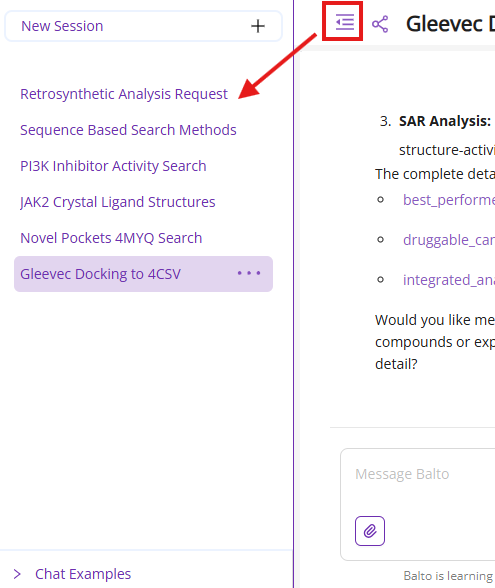
Share Button: Generates a public link to share your current chat session with colleagues

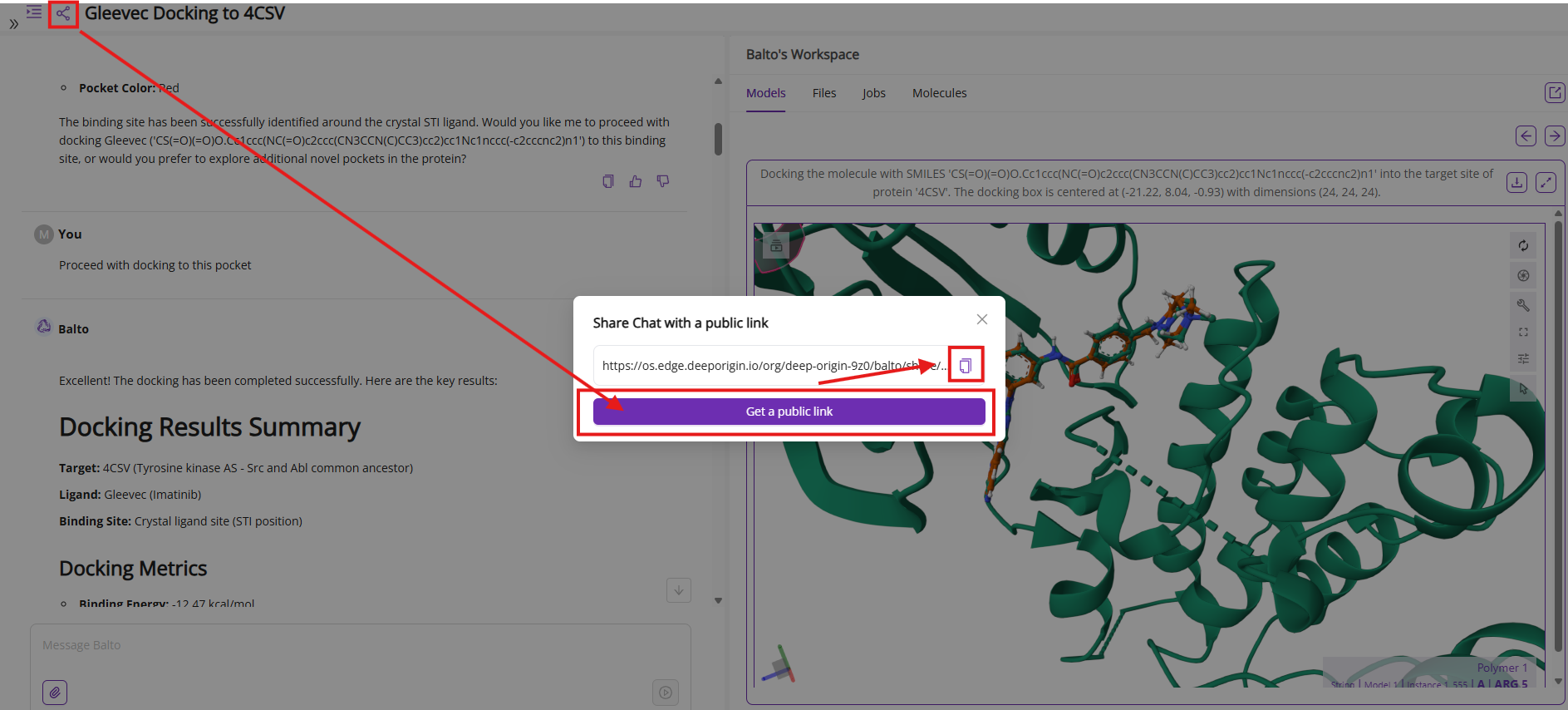
4.5 Chemical Structure Editor
You can access the 2D chemical structure editor by:
- clicking the hyperlinked SMILES string of the molecule in the molecules tab of the Balto Workspace panel opens that molecule in the editor
- clicking the Edit Molecule button in the Chat Space Panel allows you to draw a molecule from scratch or copy/paste a SMILES string into the editor to start from an existing molecule
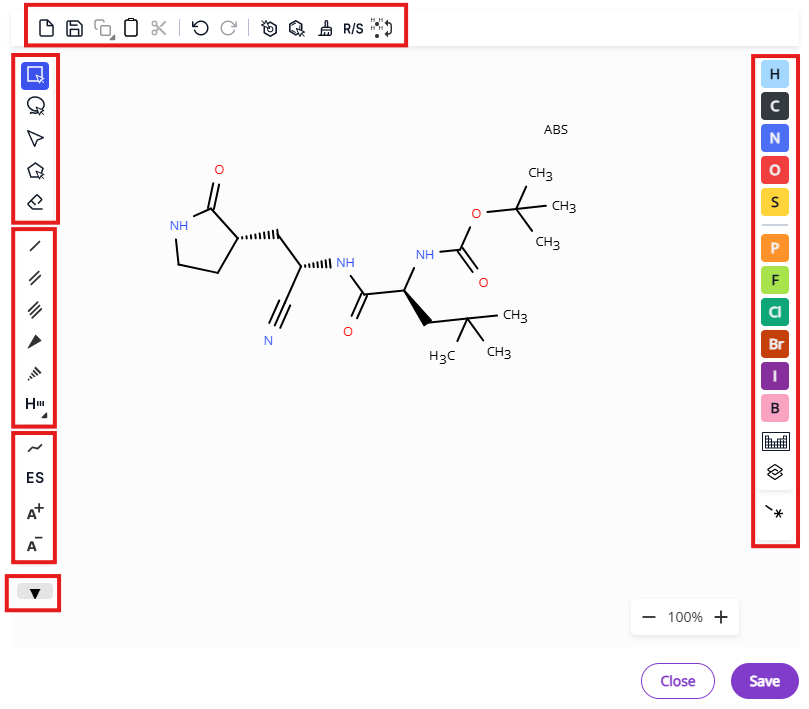
The editor contains necessary tools and several shortcuts for common moieties to facilitate editing of molecular structures. Editor tools are grouped in several menus:
- Export and bulk editing tools: Export molecule on the canvas, undo/redo operations, aromatize/dearomatize operations, clean up structure, calculate R/S designation, add/remove explicit hydrogen.
- Selection and deletion tools: Hand tool, rectangle selection, lasso selection, fragment selection, eraser.
- Bond types: Single bond, double bond, triple bond, single bond up, single bond down, hydrogen bond.
- Additional bond types are also available under the hydrogen bond expansion menu - aromatic bond, dative bond, any bond, undefined single bond, undefined double bond.
- Charges and stereochemistry: Chain tool, advanced stereochemistry, positive charge, negative charge.
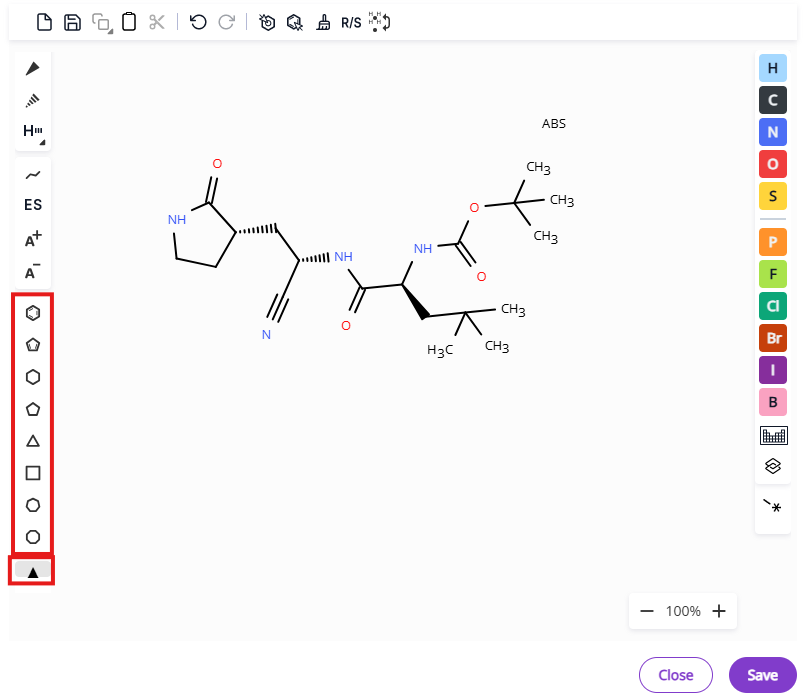
- Common rings: Benzene, cyclopentadiene and three- to eight-membered aliphatic rings.
- Atom types: Common atoms and periodic table
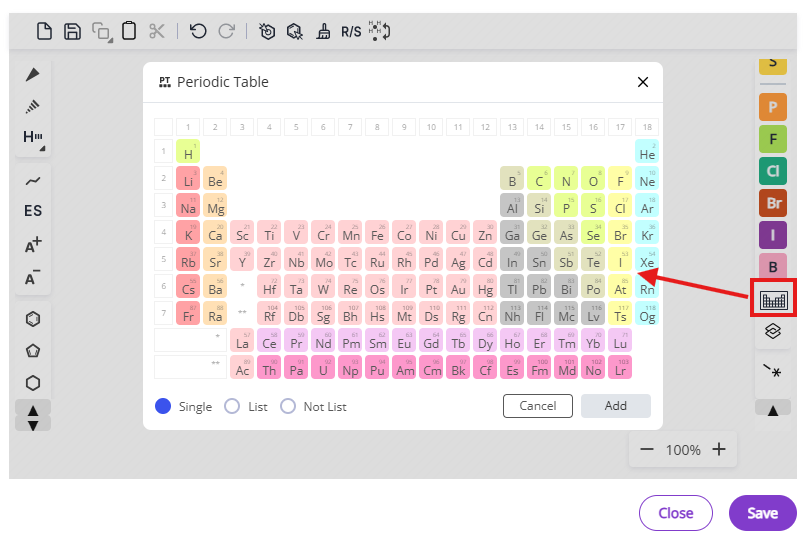
List of available shortcuts is accessible via the shortcuts button and also below:
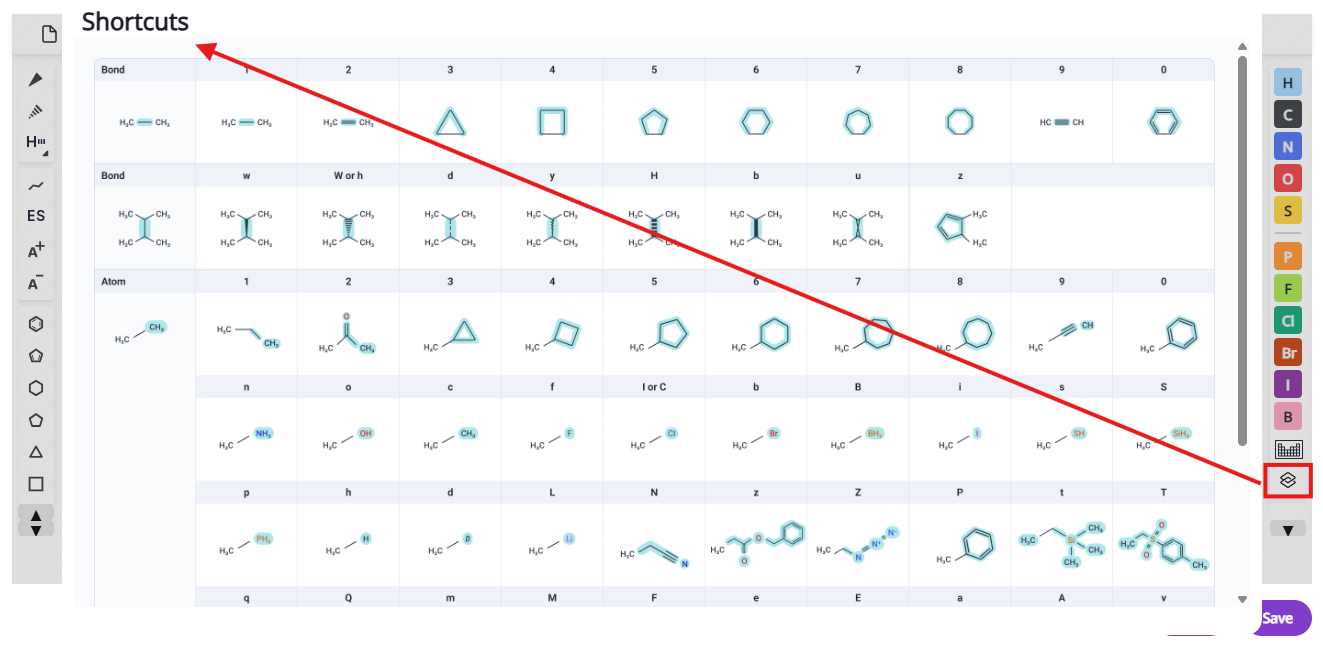
Clicking on the “Save” button will the molecule to the chat space as a SMILES string. To add the new molecule to the molecules table in the Balto Workspace panel, ask Balto to add the molecule in the chat with the SMILES string. The molecules table is a list of all the molecules gathered or uploaded for that chat, but it is not accessible across chats. You can copy the SMILES string from the molecules table to paste into a new chat or editor session.
Clicking on “Cancel” button will discard any changes made to the molecule.
5. Pricing
Subscription and pricing model
Balto uses pay-per-use pricing. Creating an account, monthly subscription and the majority of Balto actions are FREE, however Balto does charge per use for some computationally intensive tools (see paid actions section).
Free actions
Every user is given a quota of free actions (see table below) for the paid tools each month, so it is free to try out new functionality each month.
| Tool | Number of Free Actions per Month |
|---|---|
| Docking | 30 |
| Pocket Finder | 2 |
| PDF Analysis | 50 pages |
| Solubility (LogS) | 30 |
| Lipophilicity (LogP) | 30 |
| Distribution (LogD) | 30 |
| hERG inhibition | 30 |
| DO Molecular Properties | 30 |
| Ligand Protonation | 30 |
| System Prep | 30 |
| Deep Research Mode | 5 |
| CYP interaction | 30 |
| Ames Mutagenicity | 30 |
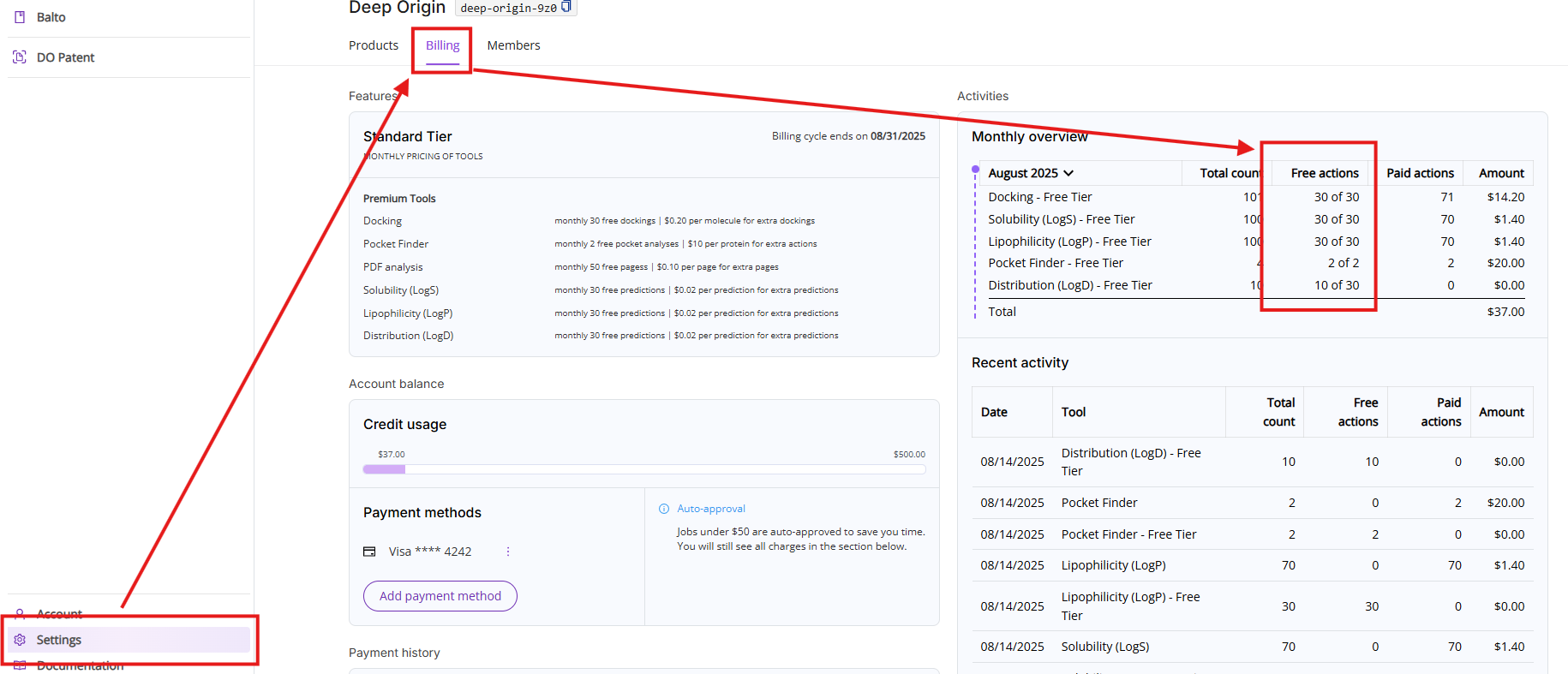
You can access remaining free actions by clicking on Settings and then on the Billing tab.
Pricing tiers
Balto has two pricing tiers:
- Standard pricing tier
- Academic pricing tier
You will automatically get an academic tier if you sign up with your .edu account.
Paid actions
Each paid tool has both an associated cost per action and a number of free actions allowed each month. Free actions are consumed first. Actions beyond the free actions limit will charged according to the table below. If you would like to adjust your pricing tier or discuss additional pricing options, then please contact support.
| Paid Tool | Number of Free Actions per Month | Academic Tier Price per Action | Standard Tier Price per Action |
|---|---|---|---|
| Docking | 30 | $0.10 per molecule | $0.20 per molecule |
| Pocket Finder | 2 | $5 per protein | $10 per protein |
| PDF Analysis | 50 pages | $0.05 per page | $0.10 per page |
| Solubility (LogS) | 30 | $0.01 per molecule | $0.02 per molecule |
| Lipophilicity (LogP) | 30 | $0.01 per molecule | $0.02 per molecule |
| Distribution (LogD) | 30 | $0.01 per molecule | $0.02 per molecule |
| hERG activity | 30 | $0.01 per molecule | $0.02 per molecule |
| DO Molecular Properties | 30 | $0.07 per molecule | $0.14 per molecule |
| Ligand Protonation | 30 | $0.01 per molecule | $0.02 per molecule |
| System Prep | 30 | $0.01 per molecule | $0.02 per molecule |
| Deep Research Mode | 5 | $5.00 per report | $5.00 per report |
| CYP interaction | 30 | $0.01 per molecule | $0.02 per molecule |
| Ames Mutagenicity | 30 | $0.01 per molecule | $0.02 per molecule |
You can always review your aggregated tool usage as well as specific actions breakdown by clicking on Settings and then on the Billing tab.
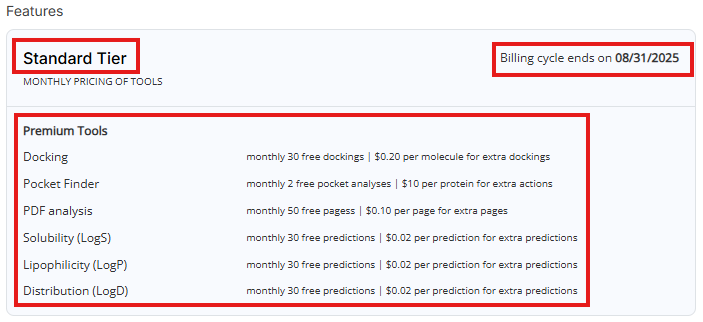
Auto-approval
Balto has a default auto-approve threshold set at $50.
Balto will automatically execute any actions that will cost less than $50 and will ask for permission to proceed if the job will cost more than $50.
Billing cycle
The credit card on file will be charged at the end of the month for any paid tool actions performed that month.
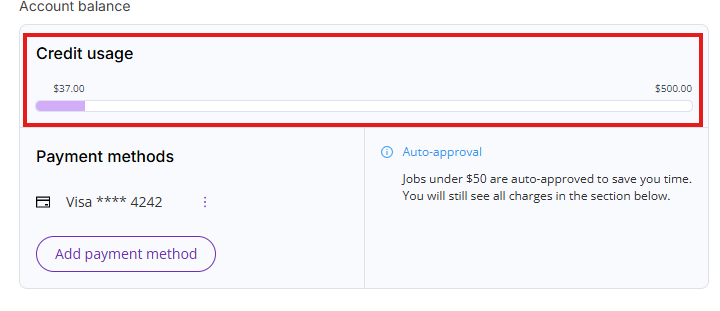
Credit usage
You will receive an automatic $500 credit when you enter a payment method. This credit allows you to use computationally intensive actions in access of monthly free actions allowance.
The mechanics of this credit limit is similar to the credit card. Credit usage accumulates all your unpaid charges for the current month (billing cycle) and unpaid charges for the previous month (billing cycle). Once the bill for the previous billing cycle is paid, your credit usage will be lowered by the paid bill amount. Once you have hit your limit for paid actions, you will not be able to perform additional paid actions without contacting Support.
You can always access your credit usage limit by clicking on Settings and then on the Billing tab.
Payments via Purchase Order
Please, contact our customer support team for this request.
6. Settings Menu
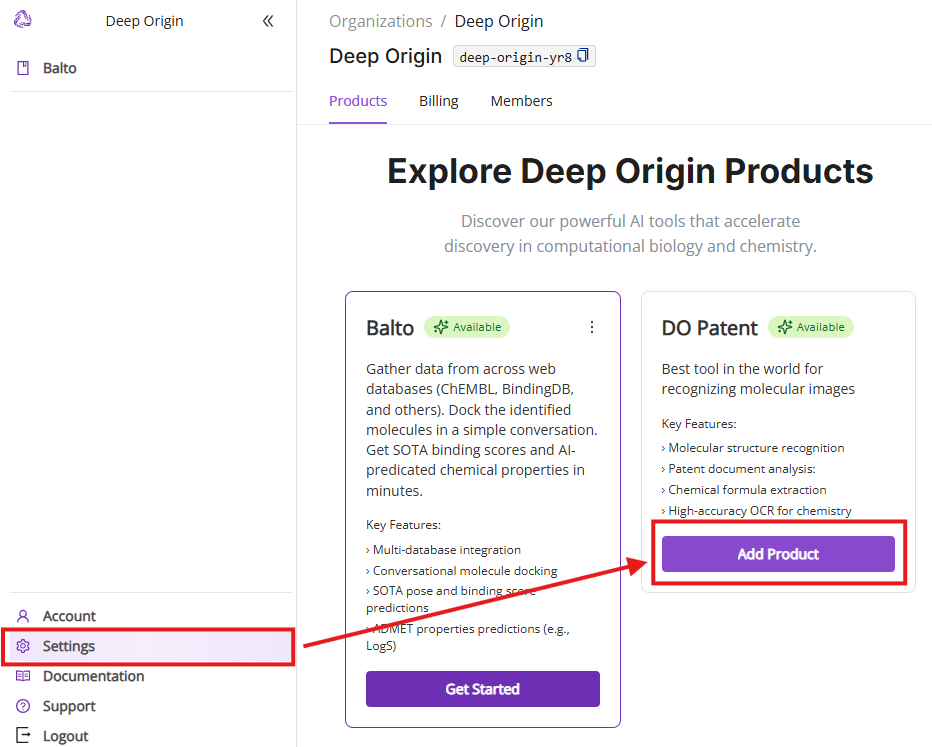
6.1 Products
Clicking “Settings” in the left navigation panel takes you to the Products tab. You can see all available Deep Origin products and add products to your product list. Subscription to additional products is FREE.
6.2 Billing tab
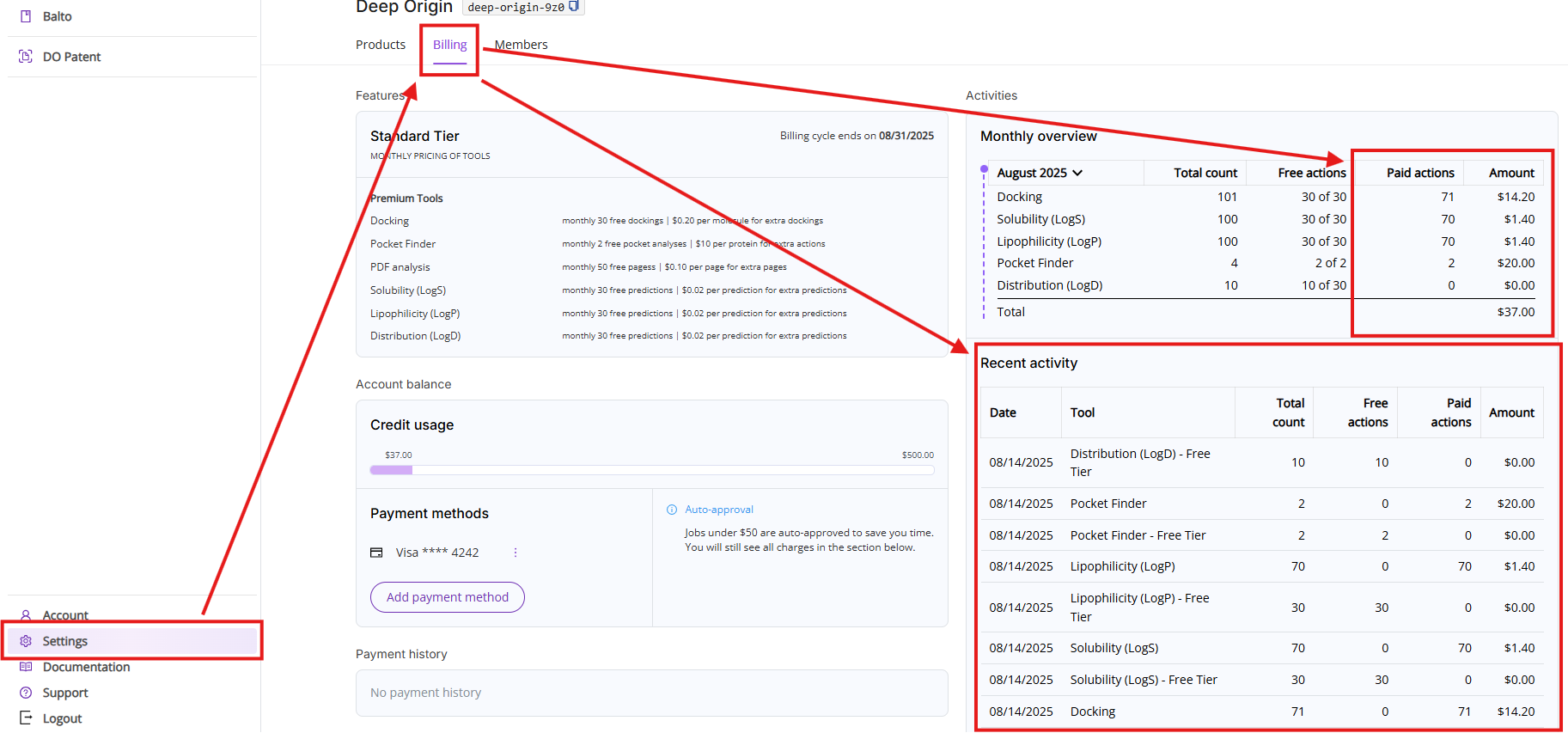
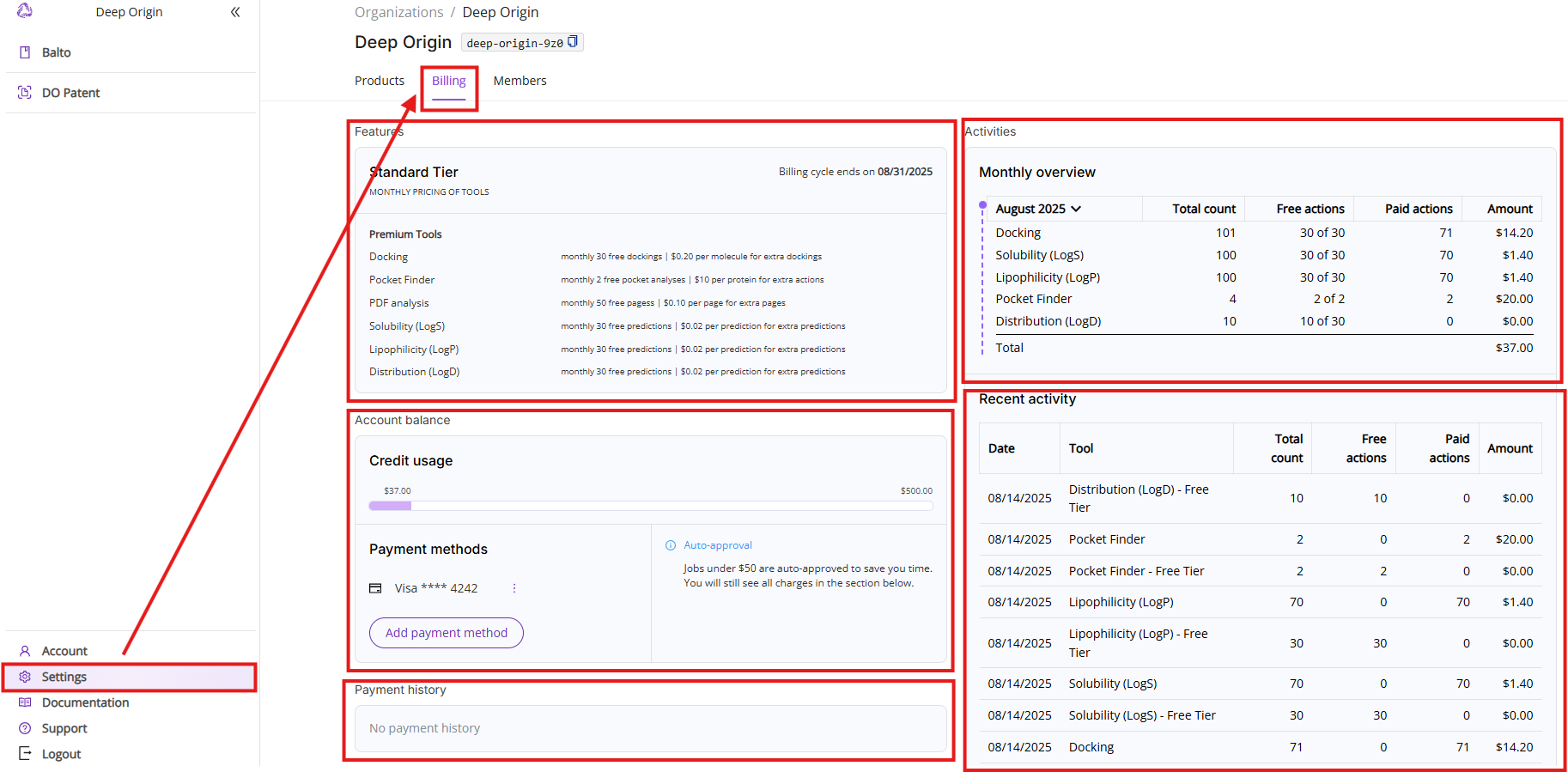
The Billing tab shows details about your current subscription, available tools, current charges and tool usage. The view consists of five sections:
- Features: Shows your current pricing tier, billing cycle and lists available paid tools with free actions limit and pricing
- Account balance: Shows current credit usage, current payment method (if one is set up) and auto-approval threshold (if payment method is set up)
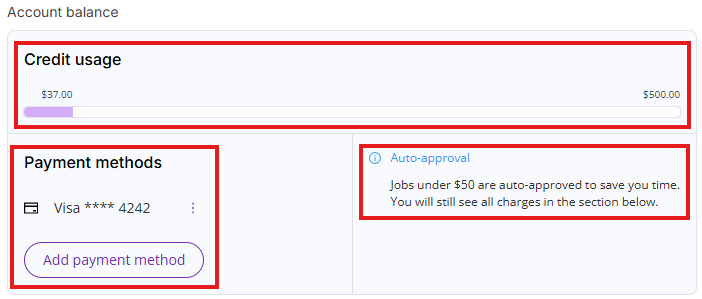
- Payment history: A table displaying past invoices for paid tool usage
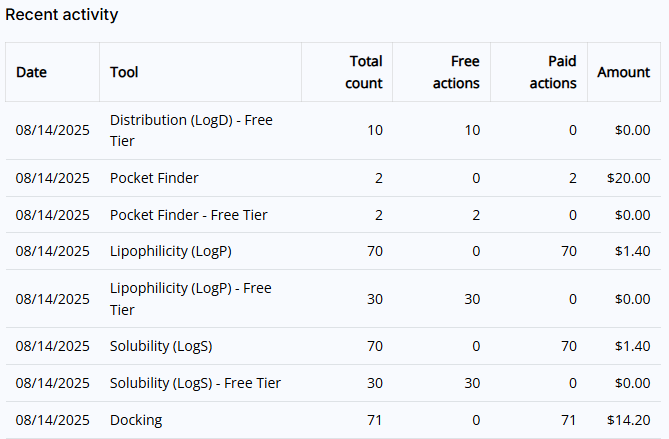
- Monthly overview: Lists all actions executed during the current month. The view is broken down into five columns:
- Period and tool names: You can select a different period by clicking on the month. Note, if a particular tool was not used during this month, it will not show up on this list.
- Total count: Shows total count of free and paid actions
- Free actions: Shows total count of used free actions and available limit of free actions in the format of XX used of YY available
- Paid actions: Shows total count of actions above the free actions limit
- Amount: Shows charges calculated by multiplying your paid actions count and the price of action

- Recent activities: Shows you a list of the last 10 executed Premium tools actions
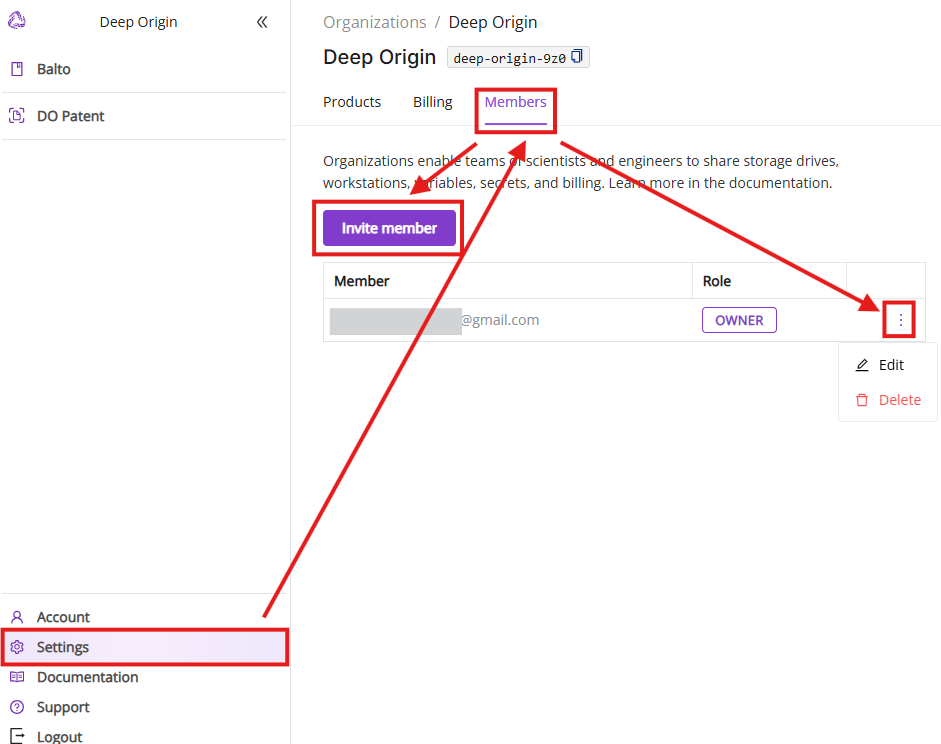
6.3 Members tab
The Members tab allows to manage your Deep Origin Organization
- Members list: Current members of your organization and their roles (e.g., “Owner”) with optional management
- Invite member: Button to invite new members
7. Support
For additional guidance, contact us through our Support Center for assistance.
8. Example 1: Dock a ligand to the specific protein structure
In this example we will dock a known small-molecule ligand to its target protein.
The target protein in this example is human soluble guanylate cyclase (SGC). SGC is one of the gasoreceptors responsible for vasodilation. The active form of the enzyme is a heterodimer consisting of alpha-1 and beta-1 subunits (UniProt entry names, GCYA1_HUMAN GCYB1_HUMAN, respectively).
We are asking to dock the ligand of interest, identified by the brand name, to the specific protein structure, identified by the PBD ID:

Balto will download the protein structure and associated information and ask you how to proceed with binding site determination. You can either select:
- A crystal ligand site (most popular)
- Find all possible binding sites with Deep Origin’s proprietary PocketFinder (most comprehensive)
- Enter amino-acid residue number(s) (e.g., 92, 145), the geometric center of which will be used for docking grid box placement
- Enter x, y, z coordinates (e.g., 1.8, 23.4, 36.2) for the docking grid box center
Note: PocketFinder is one of the paid premium tools that allows two monthly free analyses. Before using the second PocketFinder option, ensure the relevant protein chain(s) are selected for the pocket search. This requires an understanding of the drug discovery project and/or the biological unit of the target protein (e.g., monomer, homodimer, heterodimer, etc.). If needed, Balto can separate chains before running PocketFinder. In the example below, we keep both chains as we would like to target the functional unit of the SGC heterodimer.


If you select the most comprehensive option (find all binding sites with PocketFinder), Balto will find all possible binding site and associated pocket parameters such as druggability score, volume, etc. Balto’s output will include a visual representation of the pockets it found in the protein and a table summary of pocket parameters:
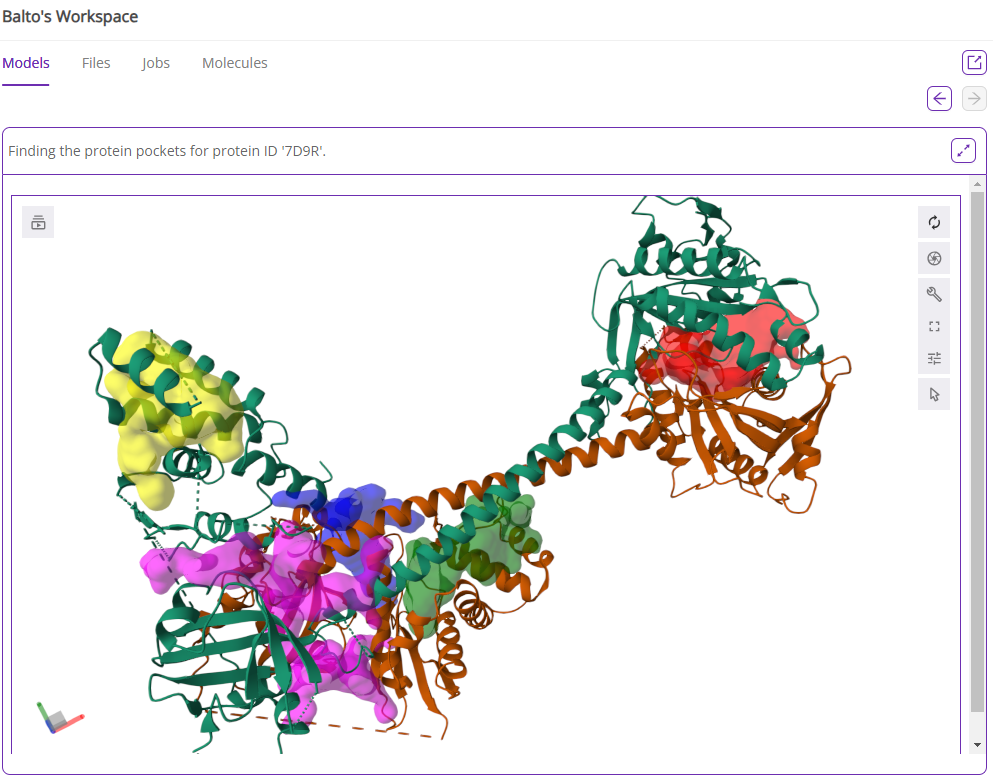
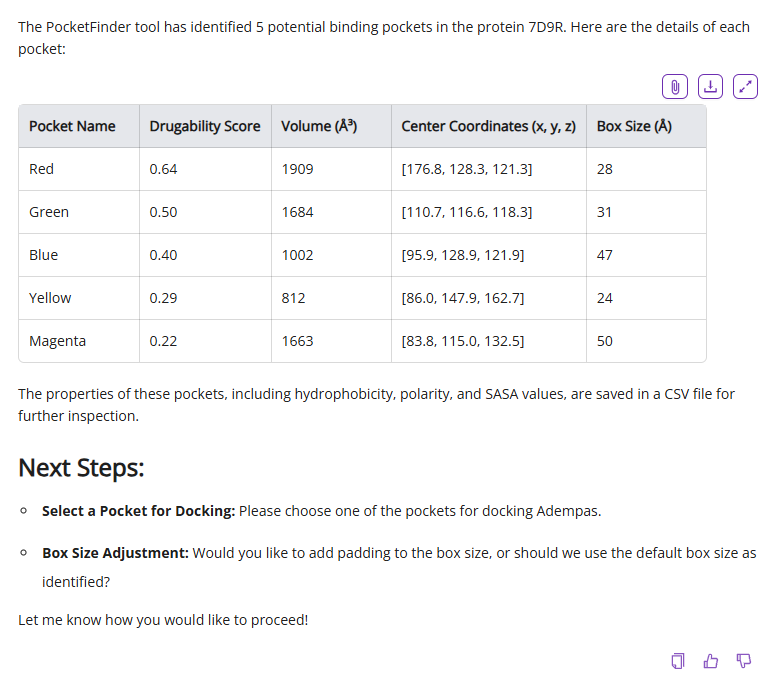
You can adjust docking box size or proceed with default parameters using one or multiple identified pockets.
Balto automatically protonates the supplied ligand at pH 7.4 and performs docking. Docking scores appear in the chat space (left), while poses are displayed in the 3D viewer (right). Click a molecule in the viewer for a close-up of the binding site. You can download the pose or expand the view to full screen using the button in the top-right corner.
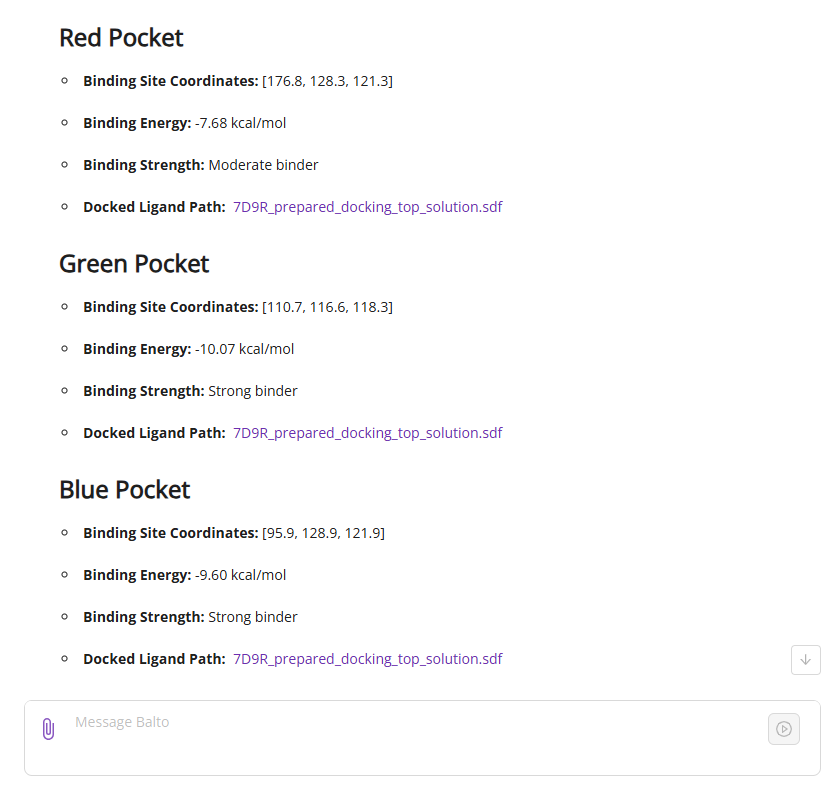

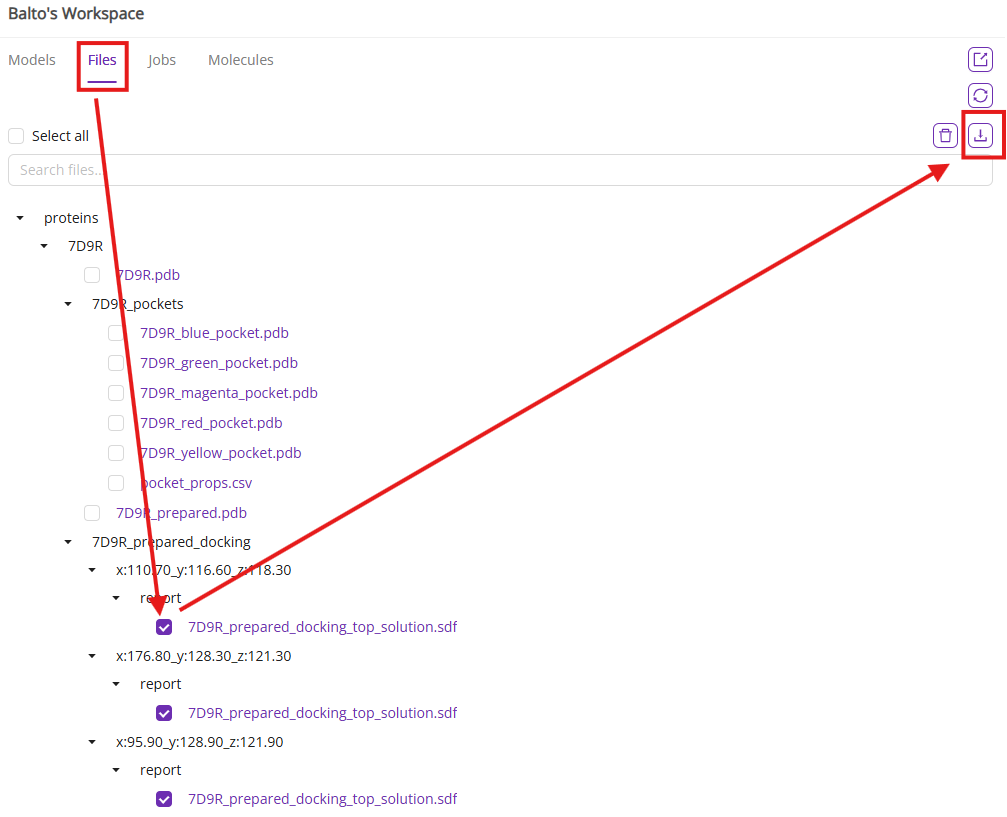
You can also export docking results in SDF format from the file system.
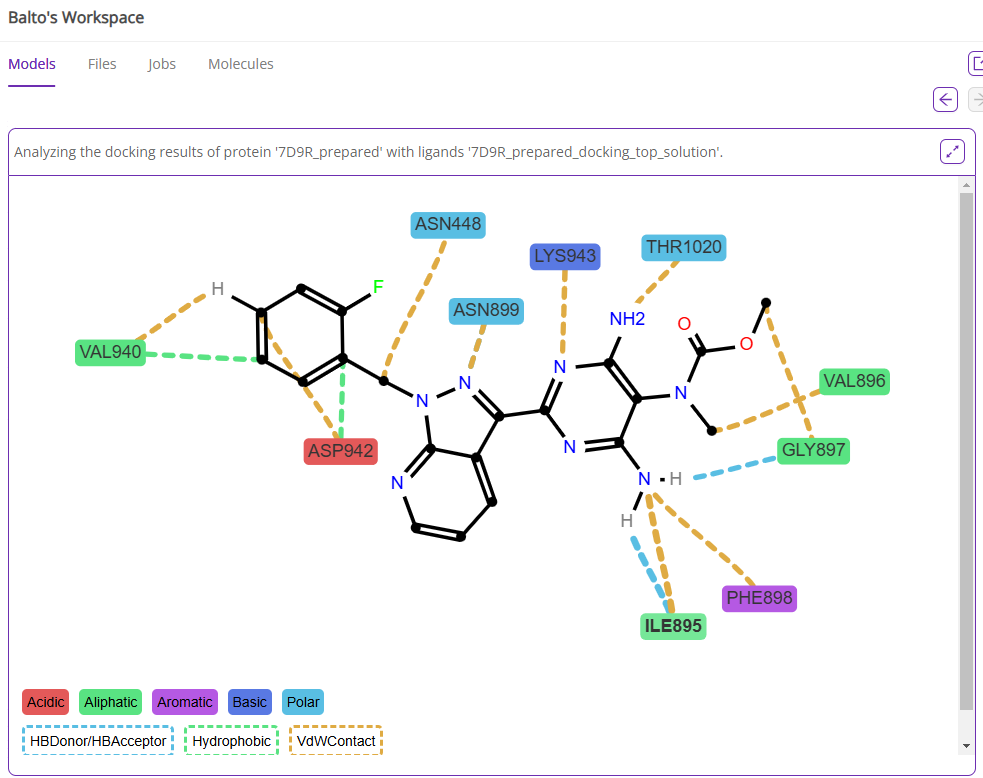
After the docking poses are available, you can execute post-docking analysis to visualize the key interactions between the ligand atoms and the binding site amino-acid residues using the integrated ProLIF method.
9. Example 2: Find a protein structure and dock a list of ligands
In this example we will select the specific protein structure and dock multiple ligands listed in a column named SMILES in a CSV file.
We will explore PI3K alpha (Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform). PIK3CA (UniPort name PK3CA_HUMAN) is an important protein in PIK3CA/MTOR/AKT signaling pathway, often targeted in oncology indications.
First, ask Balto to find relevant available structures of the PIK3CA protein. Balto will generate a sample CSV file listing 10 proteins that match the search criteria. Clicking on the file name opens a table with detailed information, including X-ray or cryo-EM resolution, protein residues, bound ligand, and its binding affinity.
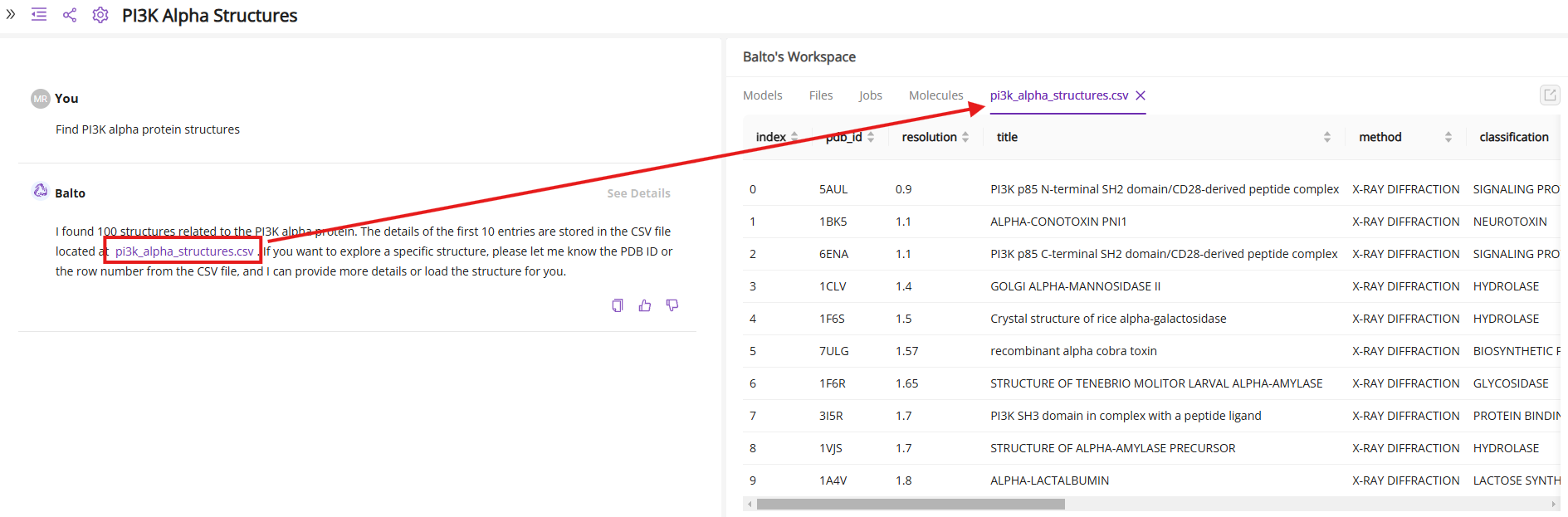
You can refine the search parameters to generate a new CSV file or request a larger list of protein structures (more than 10):
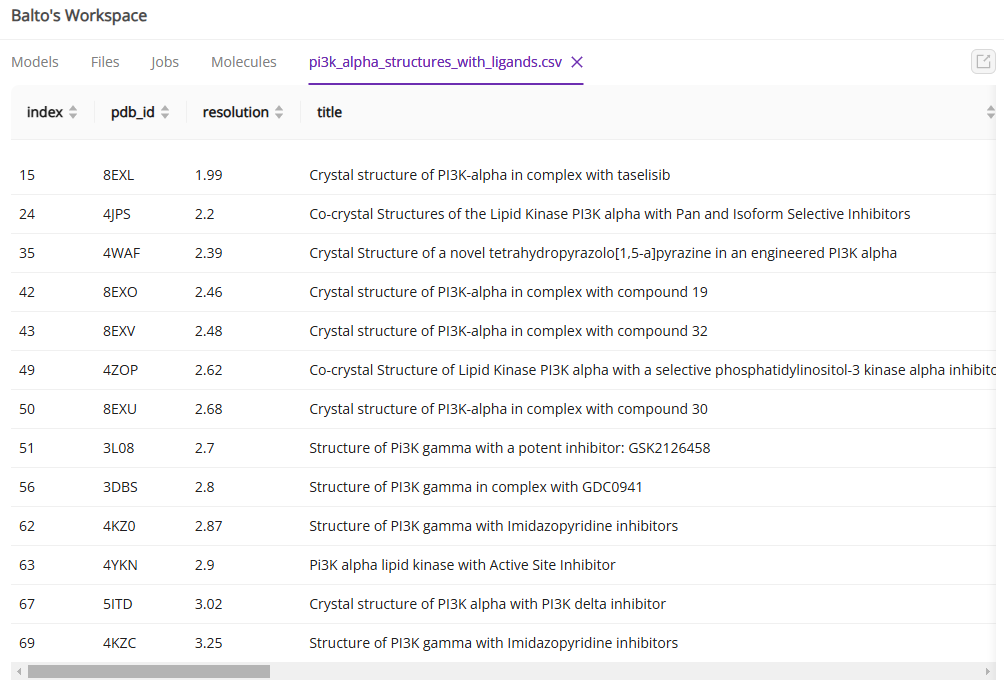
You can manually review the list of proteins and select a structure or ask Balto for recommendations:
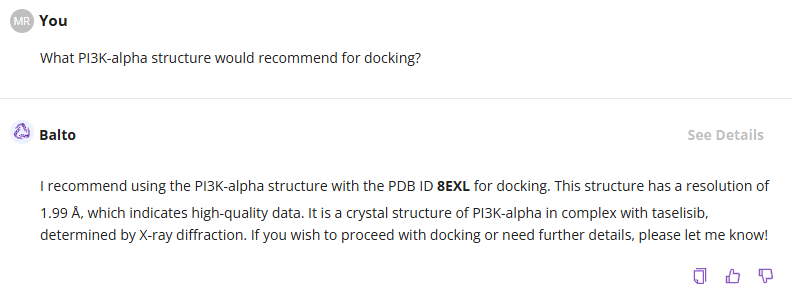
Once you selected the protein structure using steps above, you can upload your a list of ligands and ask Balto to proceed with docking.
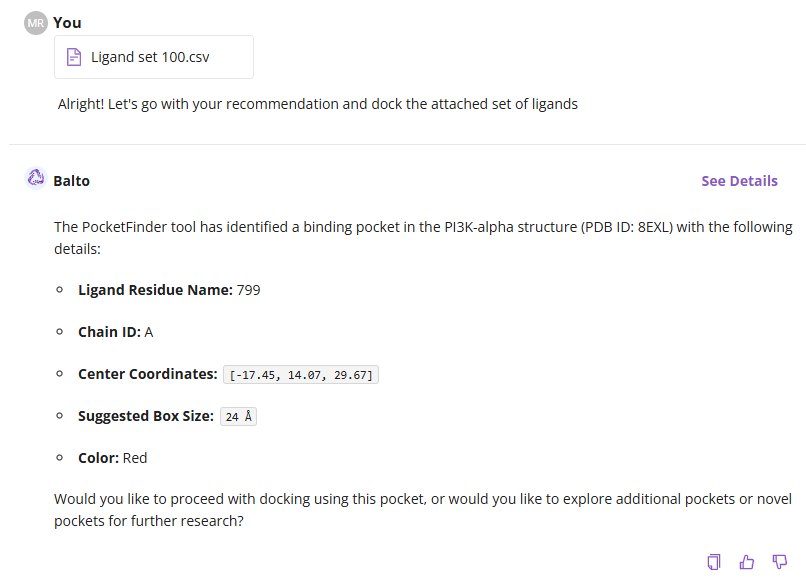
Balto will ask you to confirm the docking site. You can proceed with the crystal ligand's binding site (default) or explore additional pockets using Deep Origin’s proprietary PocketFinder (more comprehensive option).
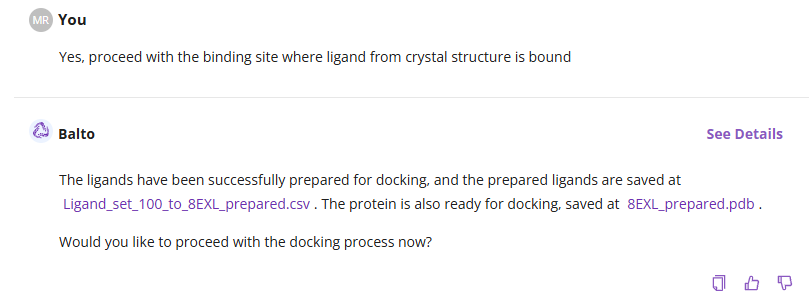
Once you confirm the binding site selection, Balto will automatically prepare the ligands for docking, including protonation state adjustments at pH=7.4. You will be asked to approve the use of credits before proceeding. If approved, Balto will begin the docking process. For ligand lists with 11 or more molecules, the task will be added to the Jobs queue and executed in the background.
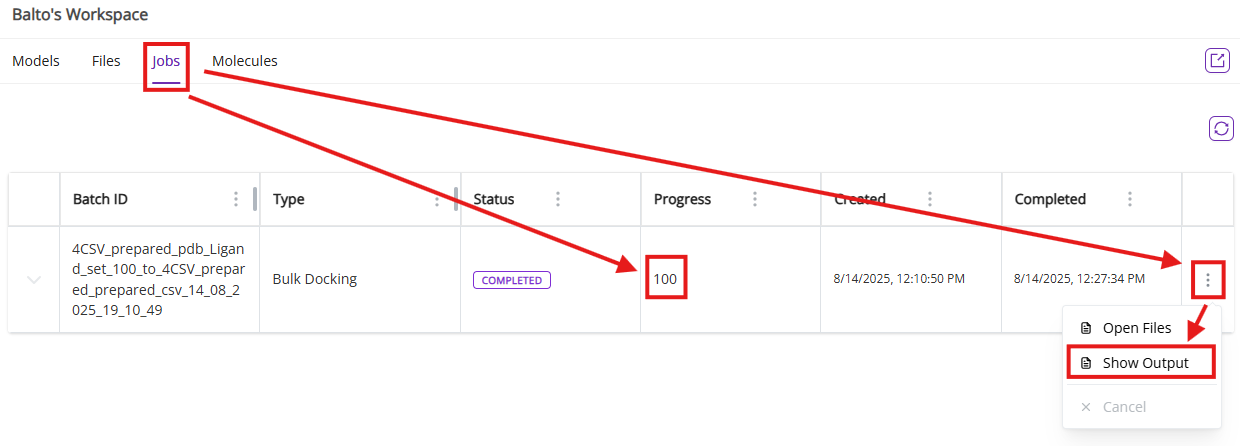
Once docking is complete, you can visualize each docked pose and overlay it with the crystal ligand by asking Balto to display docked poses. Alternatively, click the “Show models” button to review each docked pose in full-screen mode.
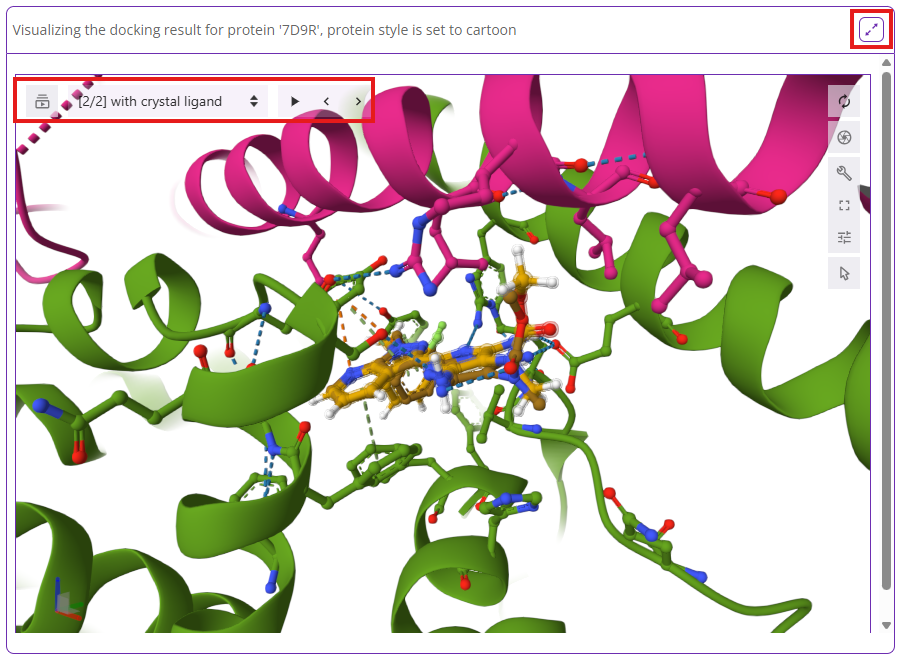
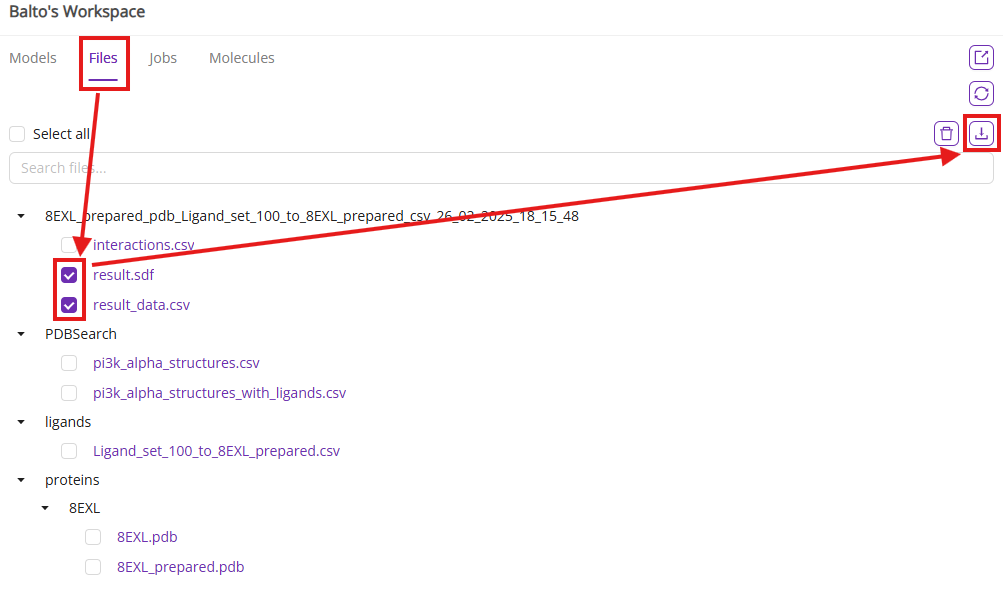
You can also download the docking results as SDF and CSV files.
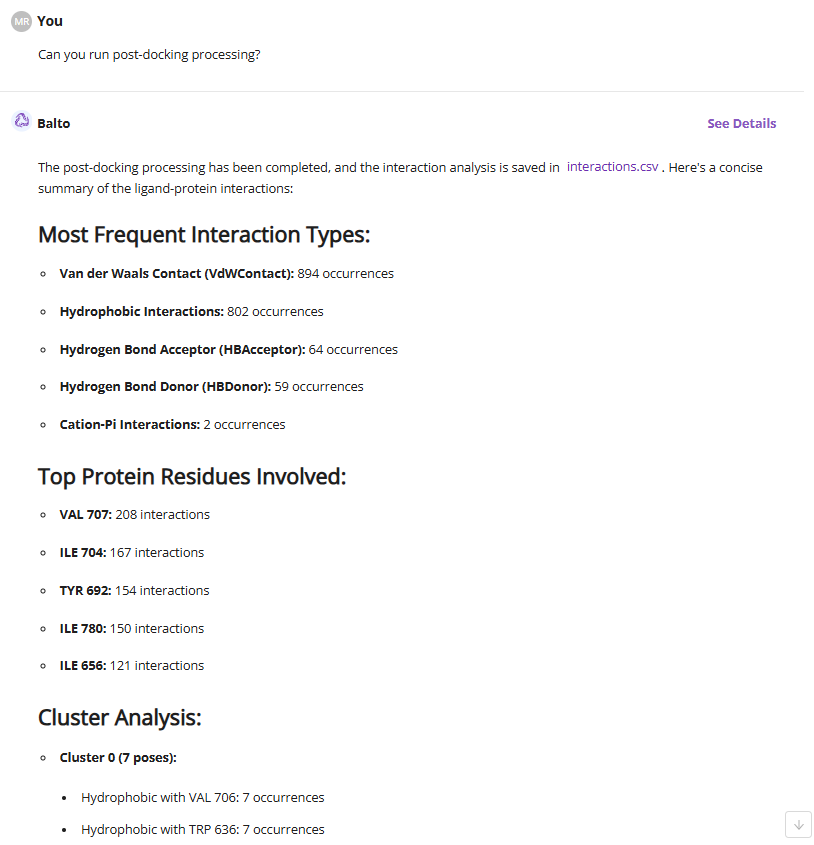
Finally, you can perform post-docking analysis to identify key protein-ligand interactions for the batch of ligands and cluster molecules by poses.
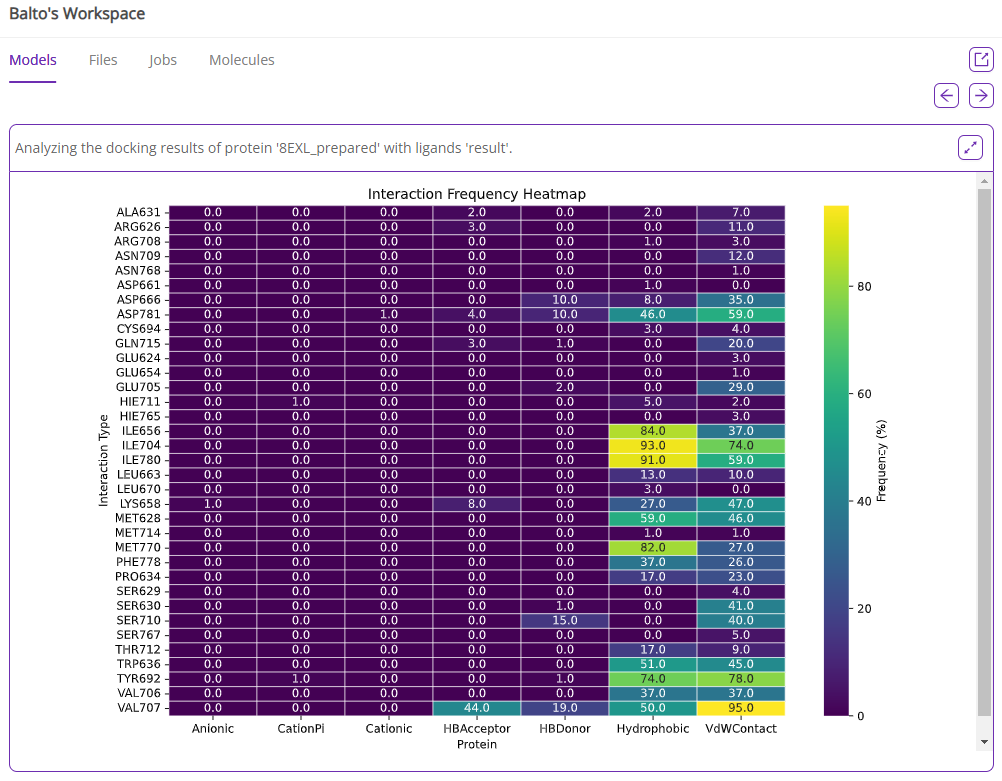
These examples demonstrate how Balto can perform complex tasks using plain English while leveraging its domain knowledge when needed. To explore additional tasks and workflows, simply ask Balto for guidance.
Happy drug hunting!